RoseTTAFold
RoseTTAFold 由华盛顿大学 David Baker 团队开发,利用深度学习技术准确、快速地预测蛋白质结构。
注意事项:
- 运行 RoseTTAFold 需要丰富的GPU资源,需要在
通用二区使用。 - 输入文件是以
.fasta格式结尾的序列文件。 - 输入文件名不能有特殊符号和
空格。 - 提交的文件第一行为标题,第二行为序列,序列为大写字母,序列之间不能有
空格或换行符。
输入文件示例:
>sequence_1
GSDNGFGSSKATSGSDFGGLAIFDGSGSEHFGHSDTHGSFDGLFGVDFZILSQQLKS
一. 模板提交
Step 1. 选择通用二区,在应用中心搜索RoseTTAFold软件,部分软件无需申请及可提交作业;
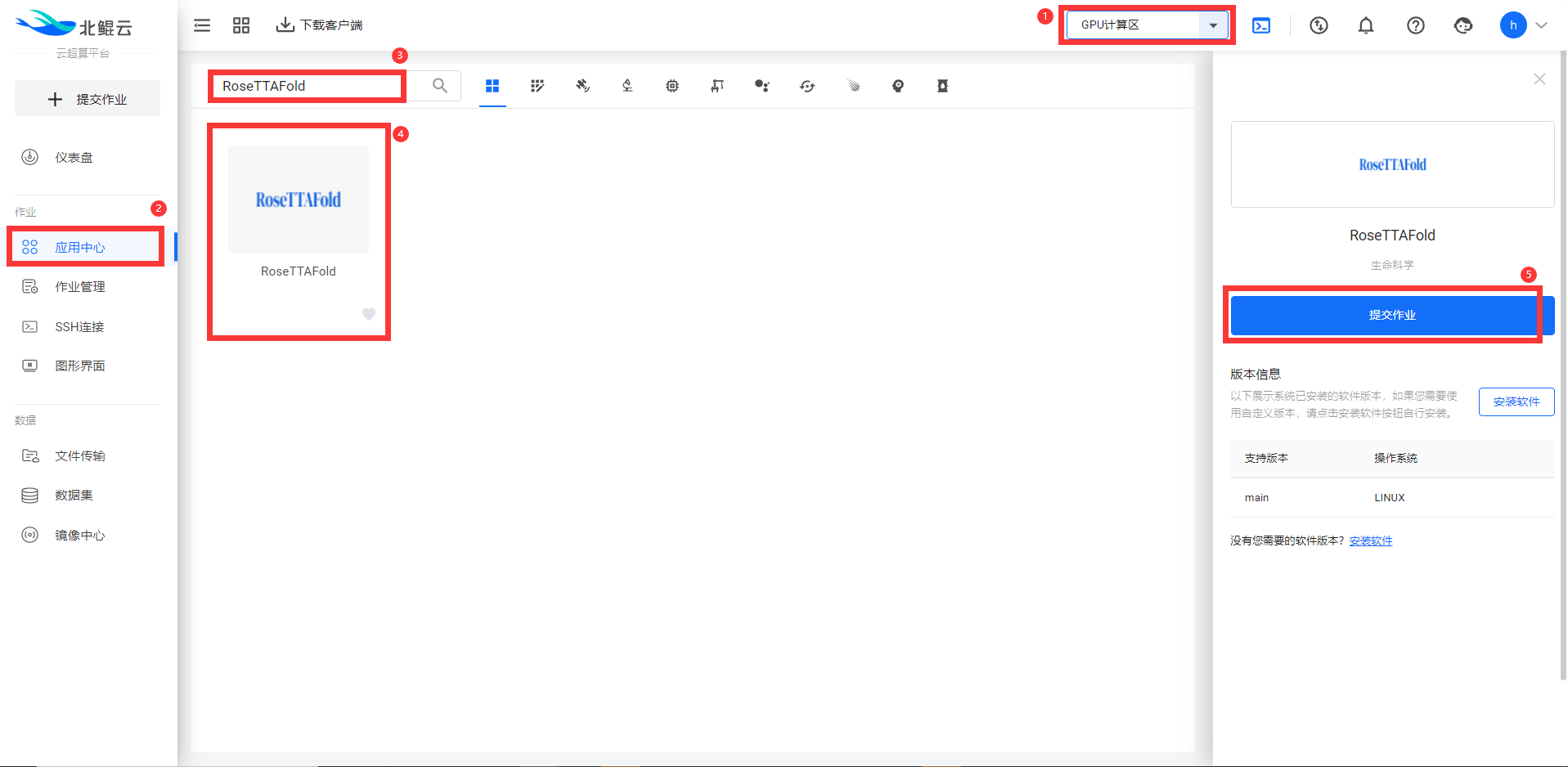
Step 2.选择可视化模板提交;

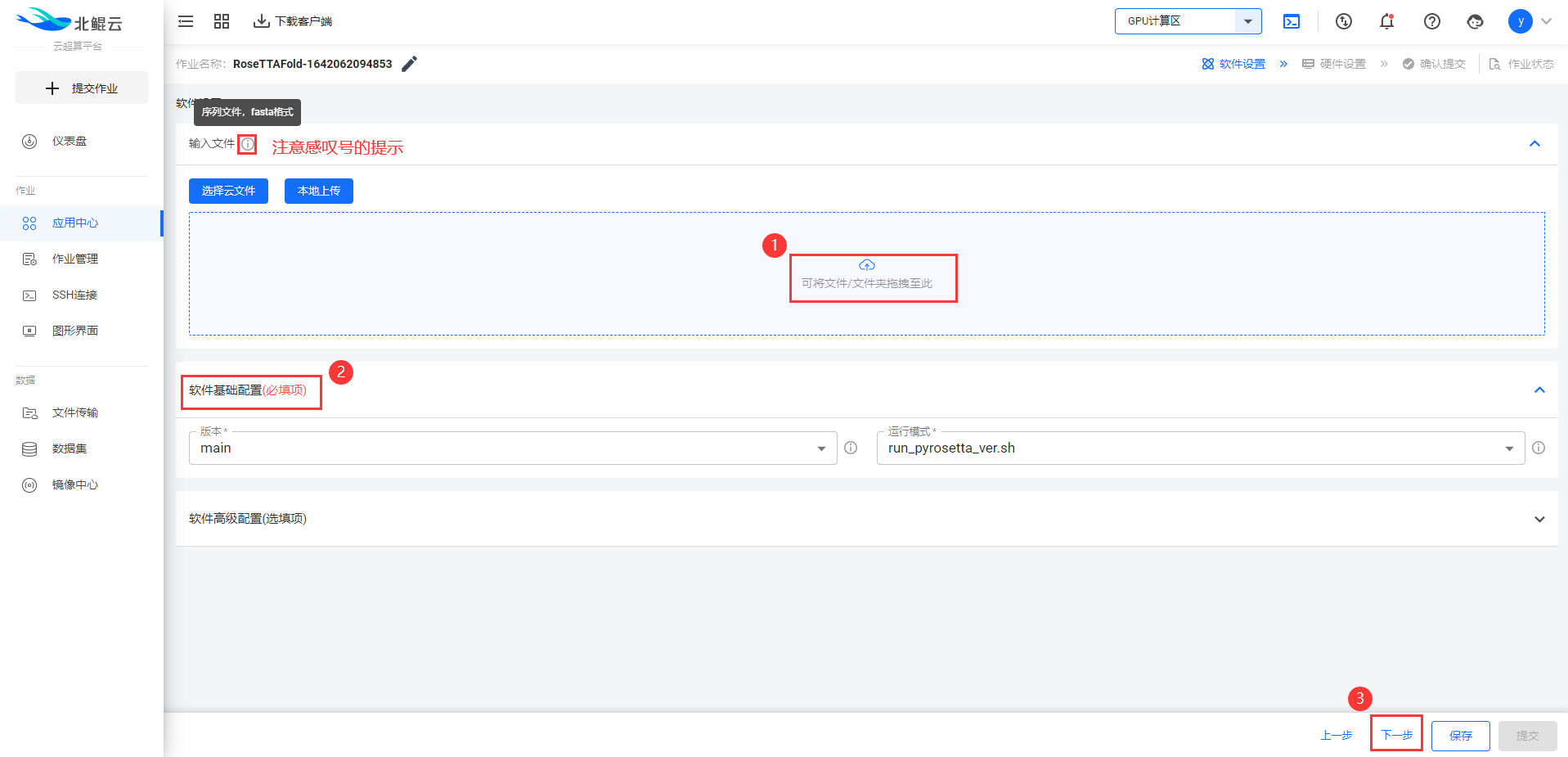
Step 3. 上传序列文件(.fasta格式),选择运行模式;
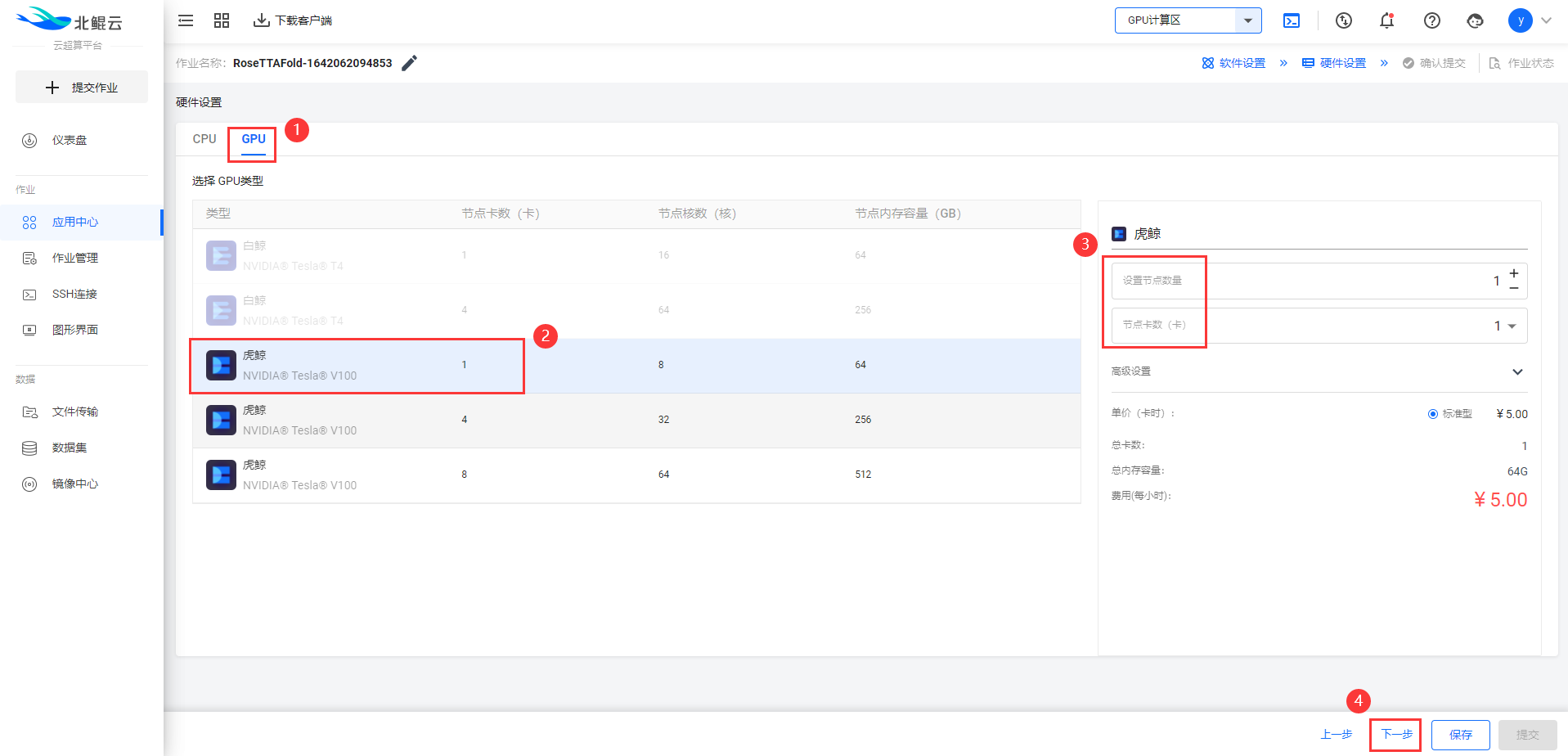
Step 4. 选择GPU硬件配置虎鲸1卡(v100-1);
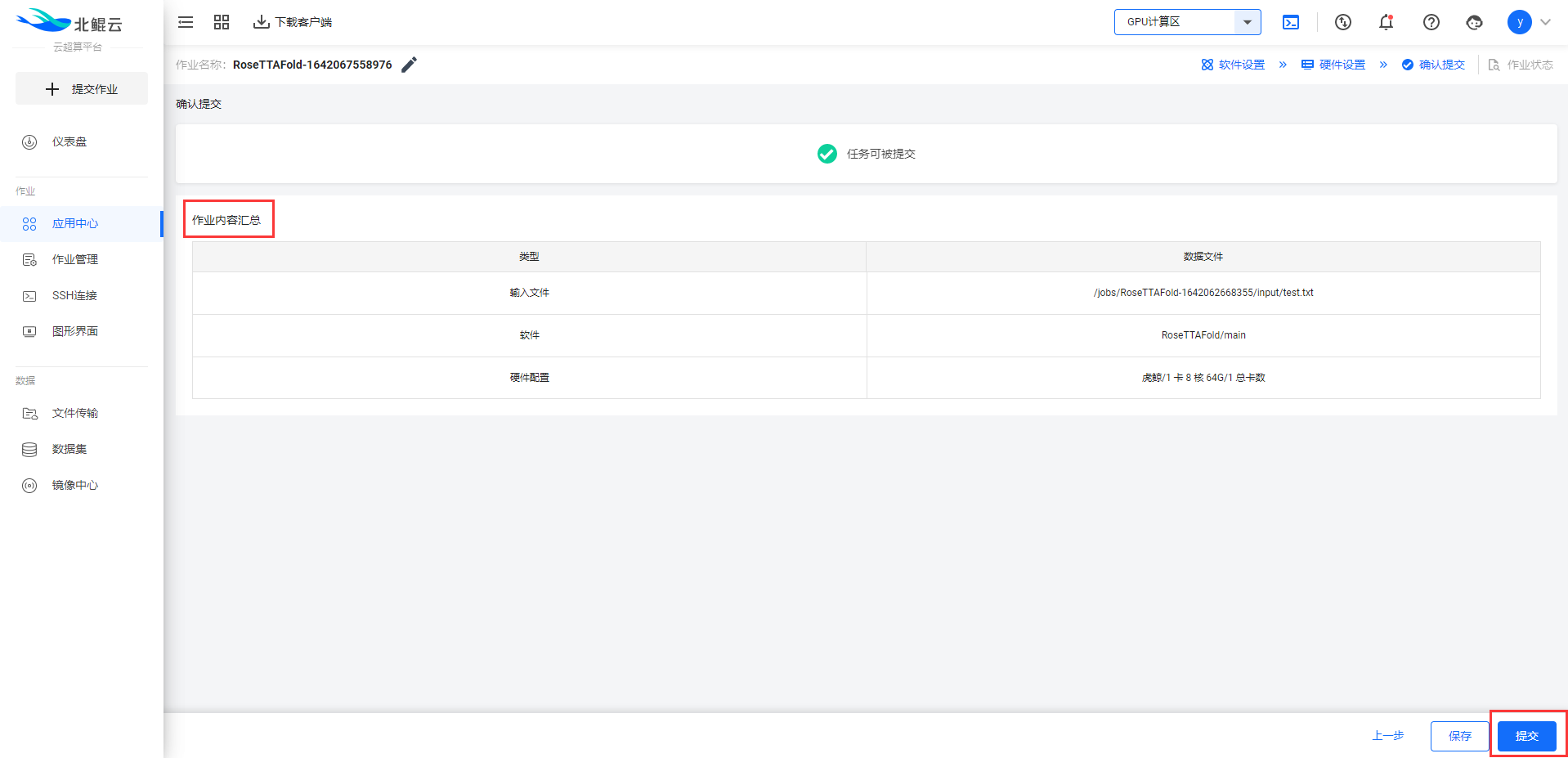
Step 5. 查看作业内容汇总,并提交作业;
Step 6. 通过作业管理查看运行中的作业;
二. 使用PyMOL对结果进行图形化展示
Step 1. 应用中心搜索PyMOL;
Step 2. 点击提交作业,选择图形界面提交;
Step 3. 启动PyMOL,选择硬件配置并启动;
Step 4. 通过VNC连接启动的linux工作站,使用软件;