标签:生命科学、动力学仿真、命令行提交
教程:GROMACS
本文提供了在北鲲云上使用GROMACS进行分子动力学模拟的分步教程
概述
GROMACS是一个用于分子动力学模拟和能量最小化的计算引擎. 分子动力学模拟和能量最小化是计算化学和分子建模领域众多技术中的两种. 计算化学 是计算技术在化学中应用, 涉及范围从分子的量子力学到复杂大分子聚集体的动力学. 分子建模 是用实际的原子模型描述复杂化学体系的一般方法, 其目的是以原子尺度的详细知识为基础, 理解和预测物质的宏观性质. 通常, 分子建模被用于设计新的材料, 因为在这过程中需要对实际体系的物理性质进行准确的预测.
本教程旨在帮助用户熟悉平台各项基础概念以及熟悉命令行提交作业的方式。通过本教程,你可以学会:
- 什么是计算区
- 如何寻找自己需要的应用
- 什么是命令行提交
- 怎么通过命令行提交超算作业
- 管理节点的若干连接方式
- 如何监控作业进度与状态
- 如何获取作业结果
大致工作流程如下:
- 创建作业目录并上传相关输入文件
- 选择GROMACS软件
- 创建集群管理节点
- slurm命令提交作业
- 作业监控
- 查看结果
step1:创建作业目录并上传相关输入文件
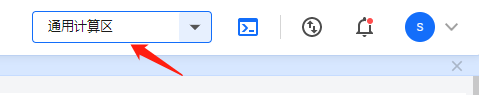
在控制台右上角选择“通用计算区”。
用户的超算作业都是在某个计算区内进行的,而不同的计算区拥有不同的硬件资源,且各有特色。所以在执行超算作业之前,根据作业实际情况选择合适的计算区是非常有必要的。若想了解各个计算区详细介绍,请点击这里。
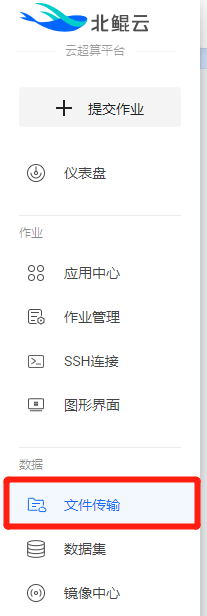
在左侧菜单栏中选择“文件传输”。
用户所有作业所需要的输入数据以及最终作业输出数据都在该页面进行统一管理。
点击“新建文件夹”按钮,在当前目录下新建“gromacsJob”文件夹。
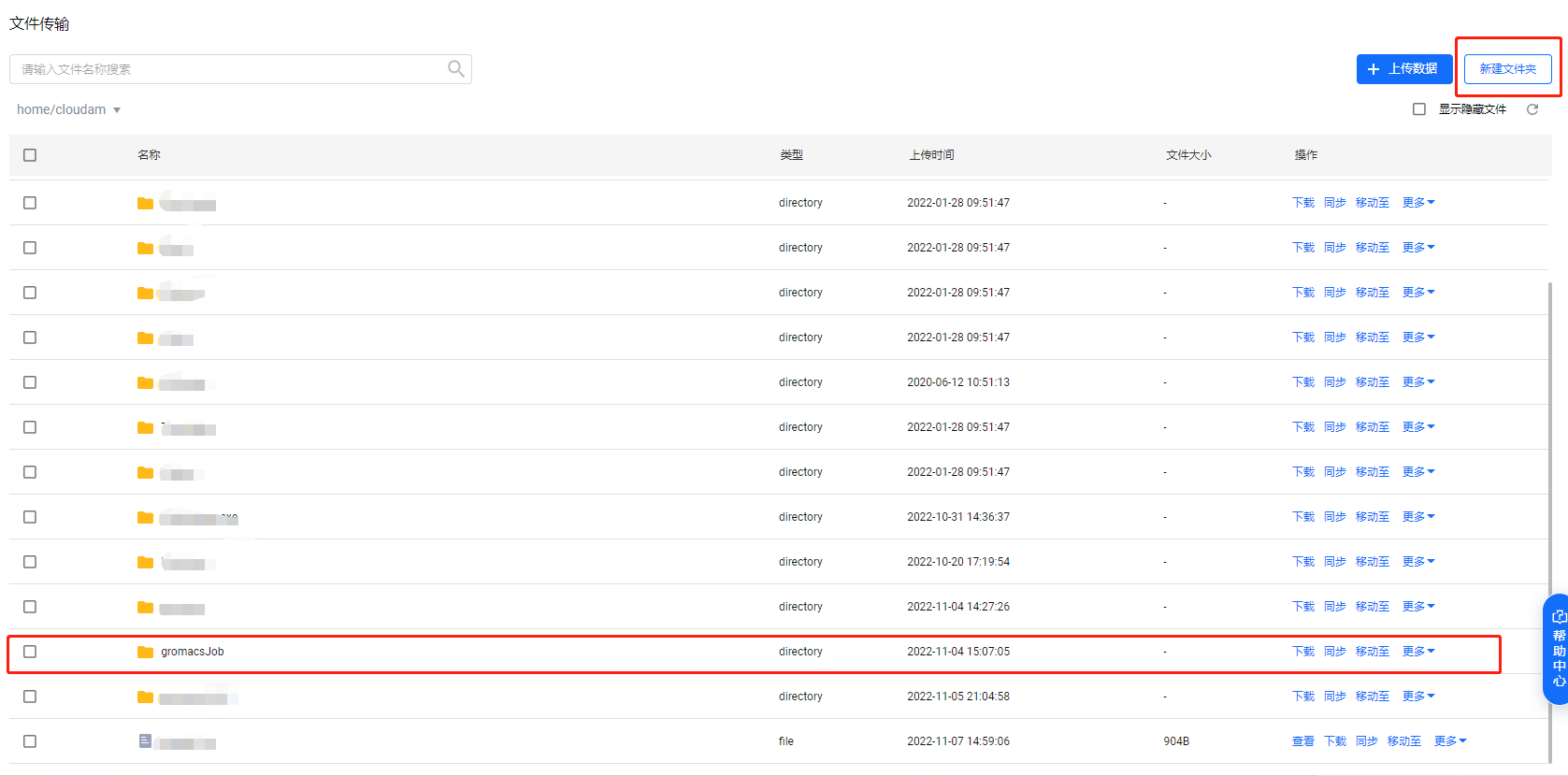
进入新建出来的“gromacsJob”文件夹内,点击“上传文件”按钮,上传
gromacs.sh和md_0_1.tpr文件。点击这里可下载本教程所用示例。
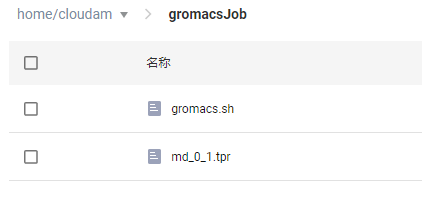
gromacs.sh是作业运行脚本文件(详见step4),md_0_1.tpr是作业所需输入文件。
step2:选择GROMACS软件
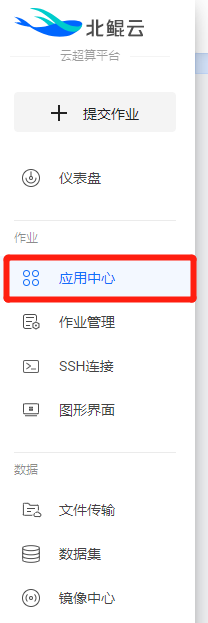
在控制台左侧菜单栏中选择并跳转至“应用中心”页面。
北鲲云平台已预装了很多软件,用户可以免安装直接使用所需软件;这些软件在“应用中心”中已按行业进行分类,各行业的热门软件/工具均已集成。若在“应用中心”没找到所需软件,可联系客服提供技术支持。
在“应用中心”页面中搜索并选中“GROMACS”软件。

在右侧弹出的软件详情栏中点击“提交作业”。
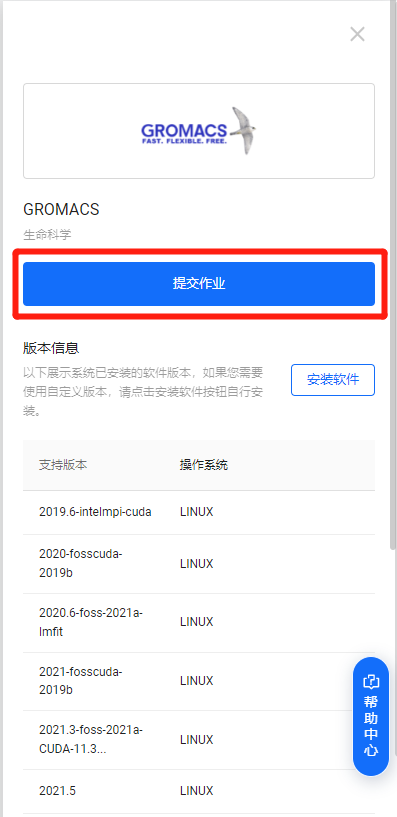
step3:创建集群管理节点
在作业提交页面,我们选择“命令行提交”。
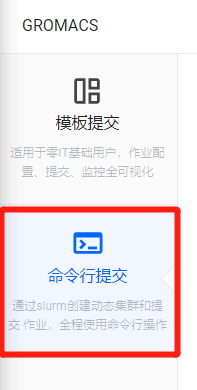
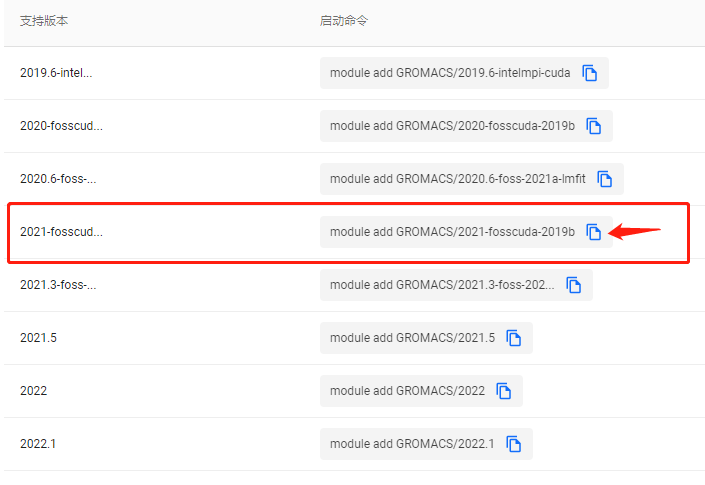
在此处我们可以查看目前平台提供的GROMACS所有版本,本教程选择使用“2021-fosscuda-2019b”版本。点击相应启动命令旁边的复制按钮可以将命令复制进粘贴板,等待后续使用。
点击“连接”。
若当前计算区内没有集群管理节点,系统将提示并帮助您创建一个;若当前计算区内已有集群管理节点,则可以选择合适的连接方式连上管理节点进行后续操作。北鲲云目前提供“Web SSH连接”、“本地SSH连接”、“XShell连接”以及“MobaXterm连接”四种方式。

step4:slurm命令提交作业
cd gromacsJob使用该命令进入我们在step1中创建的作业目录。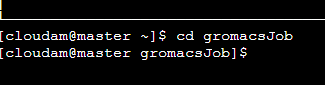
ls使用该命令可查看当前目录下的所有文件数据。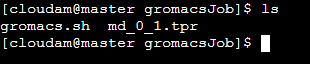
module add GROMACS/2021-fosscuda-2019b使用该命令即可加载GROMACS软件。
gmx使用该命令可以检测软件是否加载成功,当屏幕打印出以下信息后则代表GROMACS加载成功::-) GROMACS - gmx, 2021 (-:• GROMACS is written by:Andrey Alekseenko Emile Apol Rossen Apostolov• Paul Bauer Herman J.C. Berendsen Par BjelkmarChristian Blau Viacheslav Bolnykh Kevin BoydAldert van Buuren Rudi van Drunen Anton FeenstraGilles Gouaillardet Alan Gray Gerrit GroenhofAnca Hamuraru Vincent Hindriksen M. Eric IrrgangAleksei Iupinov Christoph Junghans Joe JordanDimitrios Karkoulis Peter Kasson Jiri KrausCarsten Kutzner Per Larsson Justin A. LemkulViveca Lindahl Magnus Lundborg Erik Marklund• Pascal Merz Pieter Meulenhoff Teemu Murtola• Szilard Pall Sander Pronk Roland SchulzMichael Shirts Alexey Shvetsov Alfons SijbersPeter Tieleman Jon Vincent Teemu VirolainenChristian Wennberg Maarten Wolf Artem Zhmurov• and the project leaders:• Mark Abraham, Berk Hess, Erik Lindahl, and David van der SpoelCopyright (c) 1991-2000, University of Groningen, The Netherlands.Copyright (c) 2001-2019, The GROMACS development team atUppsala University, Stockholm University andthe Royal Institute of Technology, Sweden.check out http://www.gromacs.org for more information.GROMACS is free software; you can redistribute it and/or modify itunder the terms of the GNU Lesser General Public Licenseas published by the Free Software Foundation; either version 2.1of the License, or (at your option) any later version.GROMACS: gmx, version 2021Executable: /public/software/.local/easybuild/software/GROMACS/2021-fosscuda-2019b/bin/gmxData prefix: /public/software/.local/easybuild/software/GROMACS/2021-fosscuda-2019bWorking dir: /home/cloudamCommand line:gmxSYNOPSISgmx [-[no]h] [-[no]quiet] [-[no]version] [-[no]copyright] [-nice <int>][-[no]backup]OPTIONSOther options:-[no]h (no)Print help and quit-[no]quiet (no)Do not print common startup info or quotes-[no]version (no)Print extended version information and quit-[no]copyright (yes)Print copyright information on startup-nice <int> (19)Set the nicelevel (default depends on command)-[no]backup (yes)Write backups if output files existAdditional help is available on the following topics:commands List of available commandsselections Selection syntax and usageTo access the help, use 'gmx help <topic>'.For help on a command, use 'gmx help <command>'.GROMACS reminds you: "Creativity in science, as in art, cannot be organized. It arises spontaneously from individual talent. Well-run laboratories can foster it, but hierarchical organizations, inflexible bureaucratic rules, and mountains of futile paperwork can kill it." (Max Perutz)GROMACS加载成功后,即可通过命令行形式正常使用GROMACS了。相关命令行操作可参考GROMACS官方教程。
但我们不应该在集群管理节点内直接启动GROMACS计算作业,因为集群管理节点并不具备强力的计算功能,它仅是集群作业的管理器。因此我们需要使用slurm命令将计算作业提交至计算节点,如:
sbatch -N 1 -p g-v100-1 -c 1 gromacs.sh命令。该命令表示使用1个1卡V100机器启动1个并行计算任务,任务所用的执行脚本为gromacs.sh,脚本内容如下:xxxxxxxxxxmodule add GROMACS/2021-fosscuda-2019b #加载软件export GMX_GPU_DD_COMMS=trueexport GMX_GPU_PME_PP_COMMS=trueexport GMX_FORCE_UPDATE_DEFAULT_GPU=true#生成tpr格式输入文件,如果已有tpr格式文件则不需要写#gmx grompp -f pme.mdp -c conf.gro -p tpr_file.top -o tpr_file_name.tprgmx mdrun -v -pin on -nb gpu -bonded gpu -pme gpu -cpi md_0_1 -deffnm md_0_1
step5:作业监控
使用
squeue命令可查看当前正在运行的作业列表。使用
scontrol show job JOBID命令可查看指定ID的作业详情。JOBID为你刚刚所提交作业的ID。例:作业ID为2,则命令为scontrol show job 2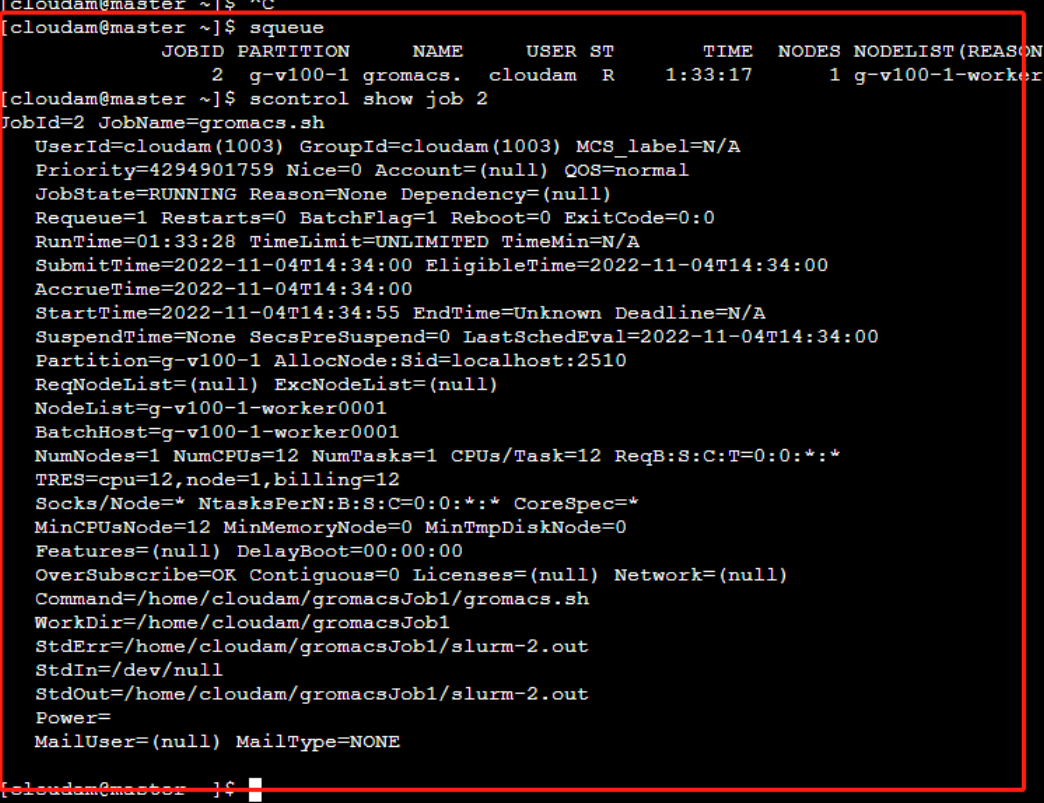
更多slurm命令可点击这里进行查看并学习。
step6:查看结果
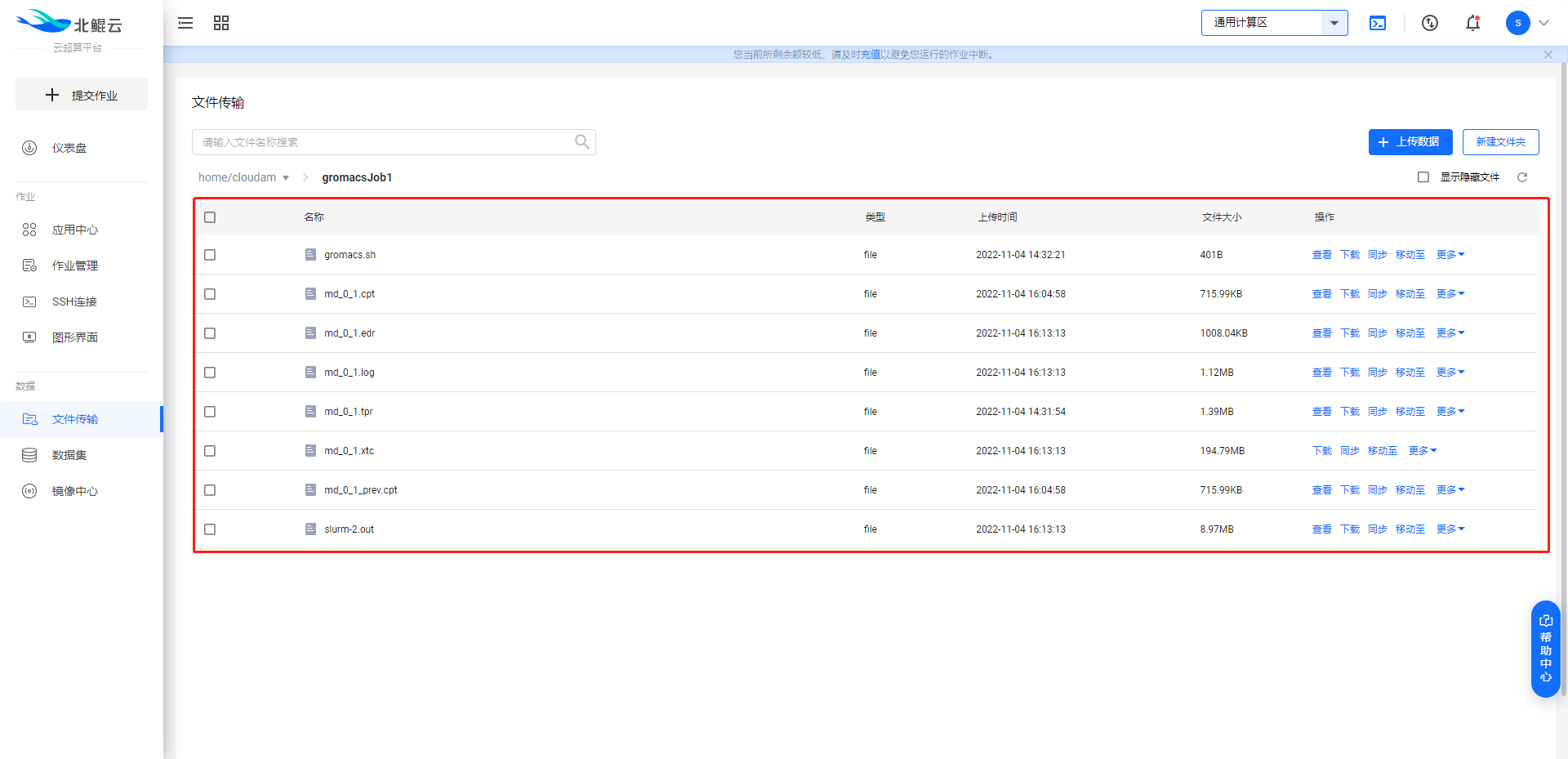
在控制台跳转至“文件传输”页面,进入step1中所建作业目录。
在该作业目录内可实时查看作业输出结果以及slurm作业日志。
相应计算结果文件可选择在线查看,也可以选择下载至本地计算机。
结语
至此,在北鲲云上使用GROMACS的整个流程就结束了。祝您体验愉快!使用过程中遇到的任何问题都可以寻找客服解决,北鲲云技术支持全天24小时为您排忧解难。
本次教程只介绍了GROMACS的简单使用,更多的GROMACS使用方法可参考手把手教你用Gromacs完成溶菌酶在水中的动力学模拟,使你更熟悉GROMACS的各项功能与操作。
若想学习使用更多的软件,可点击这里。